callvar
Overview
The callvar command is part of the MACS3 suite of tools and is used
to call variants (SNVs and small INDELs) in given peak regions from
the alignment BAM files.
Detailed Description of usage
The callvar command takes in treatment and control BAM files along
with a bed file containing peak regions. The command identifies
variants in these regions using a multi-process approach, greatly
improving the speed and efficiency of variant calling. Please check
the section Callvar Algorithm for detail on this variant calling
algorithm.
The callvar command assumes you have two types of BAM files. The
first type, what we call TREAT, is from DNA enrichment assay such as
ChIP-seq or ATAC-seq where the DNA fragments in the sequencing library
are enriched in certain genomics regions with potential allele biases;
the second type, called CTRL for control, is from genomic assay in
which the DNA enrichment is less biased in multiploid chromosomes and
more uniform across the whole genome (the later one is optional). In
order to run callvar, please sort (by coordinates) and index the BAM
files.
Example:
Sort the BAM file:
$ samtools sort TREAT.bam -o TREAT_sorted.bam$ samtools sort CTRL.bam -o CTRL_sorted.bamIndex the BAM file:
$ samtools index TREAT_sorted.bam$ samtools index CTRL_sorted.bamMake sure .bai files are available:
$ ls TREAT_sorted.bam.bai$ ls CTRL_sorted.bam.bai
To call variants:
$ macs3 callvar -b peaks.bed -t TREAT_sorted.bam -c CTRL_sorted.bam -o peaks.vcf
Command Line Options
Here is a brief overview of these options:
Input files Options:
-bor--peak: The peak regions in BED format, sorted by coordinates. This option is required.-tor--treatment: The ChIP-seq/ATAC-seq treatment file in BAM format, sorted by coordinates. Make sure the .bai file is avaiable in the same directory. This option is required.-cor--control: Optional control file in BAM format, sorted by coordinates. Make sure the .bai file is avaiable in the same directory.
Output Options:
--outdir: The directory for all output files to be written to. Default: writes output files to the current working directory.-oor--ofile: The output VCF file name. Please check the section Customized fields in VCF section for detail.--verbose: Set the verbose level of runtime messages. 0: only show critical messages, 1: show additional warning messages, 2: show process information, 3: show debug messages. DEFAULT: 2
Variant calling Options:
-gor--gq-hetero: The Genotype Quality score (-10log10((L00+L11)/(L01+L00+L11))) cutoff for Heterozygous allele type. Default is 0, or there is no cutoff on GQ.-Gor--gq-homo: The Genotype Quality score (-10log10((L00+L01)/(L01+L00+L11))) cutoff for Homozygous allele (not the same as reference) type. Default is 0, or there is no cutoff on GQ.-Q: The cutoff for the quality score. Only consider bases with quality score greater than this value. Default is 20, which means Q20 or 0.01 error rate.-For--fermi: The option to control when to apply local assembly through fermi-lite. By default (set as ‘auto’), whilecallvardetects any INDEL variant in a peak region, it will utilize fermi-lite to recover the actual DNA sequences to refine the read alignments. If set as ‘on’, fermi-lite will always be invoked. It can increase specificity, however sensivity and speed will be significantly lower. If set as ‘off’, fermi-lite won’t be invoked at all. If so, speed and sensitivity can be higher but specificity will be significantly lower.--fermi-overlap: The minimal overlap for fermi to initially assemble two reads. Must be between 1 and read length. A longer fermiMinOverlap is needed while read length is small (e.g. 30 for 36bp read, but 33 for 100bp read may work). Default is 30.--top2alleles-mratio: The reads for the top 2 most frequent alleles (e.g. a ref allele and an alternative allele) at a loci shouldn’t be too few comparing to total reads mapped. The minimum ratio is set by this optoin. Must be a float between 0.5 and 1. Default:0.8 which means at least 80% of reads contain the top 2 alleles.--altallele-count: The count of the alternative (non-reference) allele at a loci shouldn’t be too few. By default, we require at least two reads support the alternative allele. Default:2--max-ar: The maximum Allele-Ratio allowed while calculating likelihood for allele-specific binding. If we allow higher maxAR, we may mistakenly assign some homozygous loci as heterozygous. Default:0.95
Misc Options:
-mor--multiple-processing: The CPU used for mutliple processing. Please note that, assigning more CPUs does not guarantee the process being faster. Creating too many parrallel processes need memory operations and may negate benefit from multi processing. Default: 1
Example Usage
Here is an example of how to use the callvar command:
macs3 callvar -b peaks.bed -t treatment.bam -c control.bam -o experiment1
In this example, the program will identify variants in the
treatment.bam file relative to the control.bam file. The name of
the experiment is ‘experiment1’. All tags that pass quality filters
will be stored in a BAM file.
callvar Algorithm
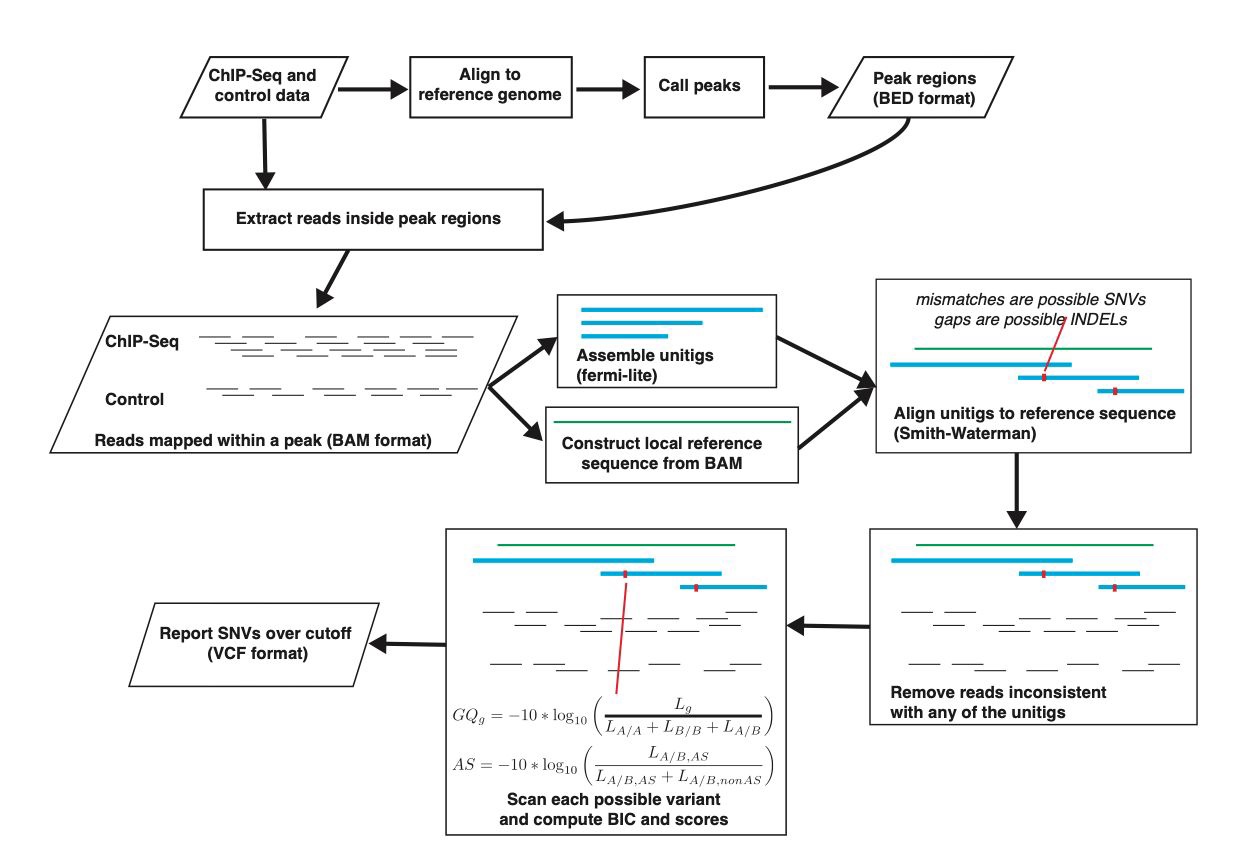
Functional sequencing assays which targeted at particular sequences, such as ChIP-Seq, were thought to be unsuitable for de novo variation predictions because their genome-wide sequencing coverage is not as uniform as Whole Genome Sequencing (WGS). However, if we aim at discovering the variations and allele usage at the targeted genomic regions, the coverage should be much higher and sufficient. We therefore proposed a novel method to call the variants directly at the called peaks by MACS3.
At each peak region, we extract the reads and assembled the DNA sequences using fermi-lite, a unitig graph based assembly algorithm developed by Heng Li. Then, we align the unitigs (i.e., assembled short DNA sequences) to the reference genome sequence using Smith-Waterman algorithm. Differences between the reference sequence and the unitigs reveal possible SNVs and INDELs. Please note that, by default, we only peform the de novo assembly using fermi-lite for detecting INDELs to save time. For each possible SNV or INDEL, we build a statistical model incorporating the sequences and sequencing errors (base qualities) from both treatment (ChIP) and control (genomic input) to predict the most likely genotype using Bayesian Information Criterion (BIC) among four allele types: homozygous loci (genotype 1/1), heterozygous loci (genotype 0/1 or 1/2) with allele bias, and heterozygous loci without allele bias. The detailed explanation of our statistical model is as follows: we retrieve the base quality scores \(\epsilon\), which represents sequencing errors, then we calculate the likelihoods of each of the four types. We assume the independence of ChIP and control experiments so that the generalized likelihood function is the product of the likelihood functions of ChIP and control data:
where \(D_c\) and \(D_i\) represent the ChIP-Seq and control (e.g., genomic input) data observed at the position including base coverage and base qualities. The parameter \(\omega\) stands for the allele ratio of allele A (chosen as the more abundant or stronger allele compared with the others) from the ChIP-Seq data and \(\phi\) represents the allele ratio in the control. The parameter \(g_c\) represents the actual number of ChIPed DNA fragments containing allele A, which could differ from the observed count \(r_{c,A}\) considering that some observations could be due to sequencing errors. The symbol \(g_i\) represents the control analogously to \(g_c\). We use \(r_c\) to denote the total number of observed allele A (\(r_{c,A}\)) and allele B (\(r_{c,B}\)). We assume the occurrence of the allele A (\(g_c\)) is from a Bernoulli trial from \(r_c\) with the allele ratio \(\omega\). The probability of observing the ChIP-Seq data at a certain position is as follows:
where \(\epsilon_j\) represents the sequencing error of the base showing difference with reference genome in case of mismatch (corresponding to SNV) and insertion. In case of deletion, the sequencing errors from the two bases on sequenced read surrounding the deletion would be considered. We model the control data in the similar way. We assess the likelihood functions of the 4 major type using the following parameters: \(\omega=1,\phi=1,g_c=r_{c,0},g_i=r_{i,0}\) for A/A genotype; \(\omega=0,\phi=0,g_c=0,g_i=0\) for B/B genotype, \(\omega=0.5,\phi=0.5\) and \(g_c,g_i\) as free variables for A/B genotype with unbiased binding; \(\phi=0.5\) and \(\omega,g_c,g_i\) as free variables for A/B genotype with biased binding or allele usage. Next, we apply the Bayesian Information Criterion (BIC) to select the best type as our prediction with the minimal BIC value among the 4 models. If the best type is either “A/B, noAS” or “A/B, AS”, we conclude that the genotype is heterozygous (A/B). We consider two types of data from the same assay independently: ChIP sample that can have biased allele usage, and control sample that won’t have biased allele usage. So that in case control is not available, such as in ATAC-Seq assay, our model can still work. Furthermore, in case a good quality WGS is available, it can be regarded as the control sample and be inserted into our calculation to further increase the sensitivity.
Customized fields in the Output VCF file
The result VCF file from MACS3 callvar will have the following
customized fields in VCF flavor:
##INFO=<ID=M,Number=.,Type=String,Description="MACS Model with minimum BIC value">
##INFO=<ID=MT,Number=.,Type=String,Description="Mutation type: SNV/Insertion/Deletion">
##INFO=<ID=DPT,Number=1,Type=Integer,Description="Depth Treatment: Read depth in ChIP-seq data">
##INFO=<ID=DPC,Number=1,Type=Integer,Description="Depth Control: Read depth in control data">
##INFO=<ID=DP1T,Number=.,Type=String,Description="Read depth of top1 allele in ChIP-seq data">
##INFO=<ID=DP2T,Number=.,Type=String,Description="Read depth of top2 allele in ChIP-seq data">
##INFO=<ID=DP1C,Number=.,Type=String,Description="Read depth of top1 allele in control data">
##INFO=<ID=DP2C,Number=.,Type=String,Description="Read depth of top2 allele in control data">
##INFO=<ID=DBIC,Number=.,Type=Float,Description="Difference of BIC of selected model vs second best alternative model">
##INFO=<ID=BICHOMOMAJOR,Number=1,Type=Integer,Description="BIC of homozygous with major allele model">
##INFO=<ID=BICHOMOMINOR,Number=1,Type=Integer,Description="BIC of homozygous with minor allele model">
##INFO=<ID=BICHETERNOAS,Number=1,Type=Integer,Description="BIC of heterozygous with no allele-specific model">
##INFO=<ID=BICHETERAS,Number=1,Type=Integer,Description="BIC of heterozygous with allele-specific model">
##INFO=<ID=AR,Number=1,Type=Float,Description="Estimated allele ratio of heterozygous with allele-specific model">
##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Read depth after filtering bad reads">
##FORMAT=<ID=GQ,Number=1,Type=Integer,Description="Genotype Quality score">
##FORMAT=<ID=PL,Number=3,Type=Integer,Description="Normalized, Phred-scaled genotype likelihoods for 00, 01, 11 genotype">