pileup
Overview
The pileup command is part of the MACS3 suite of tools and is used
to pile up alignment files. It is a fast algorithm to generate
coverage track from alignment file – either single-end or paired-end
data.
Detailed Description
The pileup command takes in one or multiple input files and produces
an output file with the piled-up genomic coverage. It uses an
efficient algorithm to pile up the alignments.
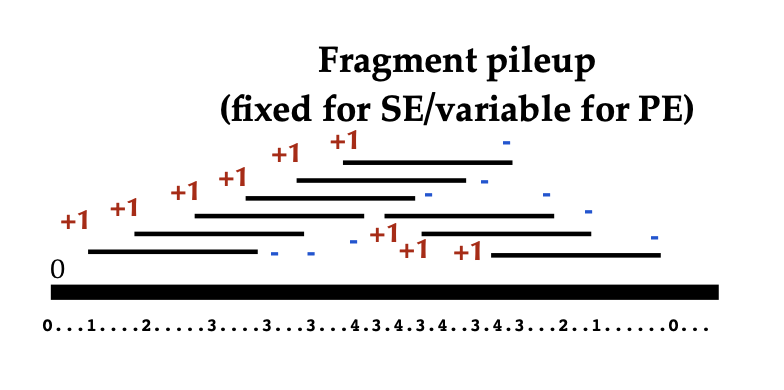
Pileup aligned reads with a given extension size (fragment size or d
in MACS language). Note there will be no step for duplicate reads
filtering or sequencing depth scaling, so you may need to do certain
pre/post-processing, such as using filterdup or randsample
command.
Command Line Options
Here is a brief overview of the command line options for pileup:
-ior--ifile: Alignment file. If multiple files are given as ‘-t A B C’, then they will all be read and combined. REQUIRED.-oor--ofile: Output bedGraph file name. If not specified, will write to standard output. REQUIRED.--outdir: If specified, all output files will be written to that directory. Default: the current working directory-for--format: Format of the tag file.AUTO: MACS3 will pick a format fromAUTO,BED,ELAND,ELANDMULTI,ELANDEXPORT,SAM,BAM, andBOWTIE. If the format isBAMPE,BEDPEorFRAG, please specify it explicitly.BAMPE,BEDPEorFRAG: When the format isBAMPE,BEDPEorFRAG, the -B and –extsize options would be ignored.
--max-count: By default, the fragment in fragment file will be counted as many as the count column indicates. For example, it will be counted twice if the count is 2. If this is not what you want, you can specify the--max-countoption, such as 1, to set a maximum count. Only usable with-f FRAG.--barcodes: Only usable with-f FRAG. This option can be used to pileup fragments only from a subset of barcodes, such as those representing a particular cluster of cells. You can provide a plain text file in which each row represents a unique barcode such as:AAACGAAAGACTCGGA AAACGAAAGTTTCGGA ...
-Bor--both-direction: By default, any read will be extended towards the downstream direction by the extension size. If this option is set, aligned reads will be extended in both upstream and downstream directions by the extension size. This option will be ignored when the format is set asBAMPE,BEDPEorFRAG. DEFAULT: False--extsize: The extension size in bps. Each alignment read will become an EXTSIZE of the fragment, then be piled up. Check description for-Bfor details. This option will be ignored when the format is set asBAMPE,BEDPEorFRAG. DEFAULT: 200--buffer-size: Buffer size for incrementally increasing the internal array size to store read alignment information. In most cases, you don’t have to change this parameter. However, if there are a large number of chromosomes/contigs/scaffolds in your alignment, it’s recommended to specify a smaller buffer size in order to decrease memory usage (but it will take longer time to read alignment files). Minimum memory requested for reading an alignment file is about # of CHROMOSOME * BUFFER_SIZE * 8 Bytes. DEFAULT: 100000--verbose: Set verbose level. 0: only show critical messages, 1: show additional warning messages, 2: show process information, 3: show debug messages. If you want to know where are the duplicate reads, use 3. DEFAULT: 2
Example Usage
Here is an example of how to use the pileup command:
macs3 pileup -i treatment.bam -o piledup.bedGraph -f BAM --extsize 147
In this example, the program will pile up the alignments in the
treatment.bam file and write the result to piledup.bedGraph. The
input file is in BAM format, and we extend each sequencing tag into a
147bps fragment for pileup.